2000-2005B: The Synaptic Signaling Network
|
Heading into this study, we knew we were on to something big. Our studies, and related studies by others, had revealed a group of signal transduction pathways that appeared fundamentally important for controlling the activity level of synapses during behaviors. The pathway components are all conserved and abundant in human brain, and many had been shown to be important for learning and memory. Our studies seemed to suggest that they were not just important for higher brain functions such as learning and memory, but that they seemed to be critical for much more basic functions of neurons, such as connecting neuronal activity with organized, coordinated behavior, possibly as fundamental as turning synapses ON and OFF.
Our study in Adventures>2000-2005A had complicated matters by identifying a third major pathway, the Gs pathway, that had be accounted for in our model. In the current Adventure, we started by separately investigating the relationship of the Gq and Gs pathways to neurotransmitter release and got some surprising results. We then investigated the relationship of the Gq and Gs pathways to each other, asking the question "How do the Gq and Gs pathways interact in the broader synaptic signaling network to coordinate the synaptic transmission that drives locomotion and neurotransmitter release in C. elegans?" Finally, we wanted to know the relationship of the RIC-8 protein, which we had discovered in 2000, to the Gq and Gs pathways. Greg Tall had just found that RIC-8 can function as a GEF for Gq, but he hadn't yet been able to show that it had the same activity for Gs. Our data from Adventures>2000-2005A hinted that RIC-8 might be important for Gs activity as well, but we needed to further test that genetically. |


|
In this study we relied heavily on a genetic technique known as "epistasis analysis". This is a powerful method which relies on the analysis of strategically-constructed double mutants to infer the upstream/ downstream order of proteins in a signaling pathway or, on a larger scale, pathways in a network.
To be successful epistasis analysis requires the right set of mutants. Fortunately, the genetic studies of ourselves and others in C. elegans had produced a remarkable set of genetic mutations and tools, unique among model organisms in their scope, that are well-suited for epistasis studies of the Go, Gq, and Gs synaptic signaling pathways. These included mutations that completely knock out each of these 3 pathways, mutations that knock out the UNC-13 synaptic vesicle priming protein hypothesized to be one important target of the pathways, transgenic strains and native gain-of-function mutants in which each pathway is strongly hyperactivated, a variety of mutations in negative regulatory components whose loss of function lead to hyperactivation of each pathway, and transgenic and pharmacological tools that allow each pathway to be acutely activated in living animals.
Adding to these tools, and facilitating their use in epistasis analysis, is the amazing ability of C. elegans mutants lacking these pathways to remain alive for long periods of time and, in some cases, to even reproduce as homozygotes under standard or specially controlled lab conditions.
We started by simply analyzing the function of the Gq pathway separate from the other pathways. Studies from the Kaplan lab and others had suggested that the synaptic vesicle priming protein UNC-13 might be a major downstream target of the DAG produced by the Gq pathway. Could the major function of the Gq pathway be to cause SV release through a DAG and UNC-13-dependent mechanism? To test this, we first asked if could bypass the EGL-30 (Gq) pathway by simply putting animals on plates containing an optimal level of a DAG analog called phorbol myristate acetate (phorbol ester). This is a very hydrophobic small molecule that soaks into the animal and into its neurons quickly.
egl-30 (Gq) null mutants arrest as tiny paralyzed larvae, but they can live for several weeks on culture plates, pumping in a small amount of bacterial food every once in a while due to the myogenic activity of the pharynx, but not growing past the second or third larval stage. This is similar to the phenotype that is seen when animals can't release neurotransmitter. However, that same phenotypes could also be caused by neuronal development defects or muscle defects that have nothing to do with the synaptic function of neurons. However, we were able to show that their paralysis and growth defects resulted from an inability to produce the DAG signal necessary for synaptic transmission and locomotion.
To show this, we simply placed the arrested egl-30 larvae on plates containing various concentrations of phorbol ester, waited 2 hours, and then observed the animals and quantified their locomotion rates. We were able to find a phorbol ester concentration that restored wild type levels of locomotion to egl-30 larvae and that allowed them to escape their larval arrest and grow for several generations on the plates.
To be successful epistasis analysis requires the right set of mutants. Fortunately, the genetic studies of ourselves and others in C. elegans had produced a remarkable set of genetic mutations and tools, unique among model organisms in their scope, that are well-suited for epistasis studies of the Go, Gq, and Gs synaptic signaling pathways. These included mutations that completely knock out each of these 3 pathways, mutations that knock out the UNC-13 synaptic vesicle priming protein hypothesized to be one important target of the pathways, transgenic strains and native gain-of-function mutants in which each pathway is strongly hyperactivated, a variety of mutations in negative regulatory components whose loss of function lead to hyperactivation of each pathway, and transgenic and pharmacological tools that allow each pathway to be acutely activated in living animals.
Adding to these tools, and facilitating their use in epistasis analysis, is the amazing ability of C. elegans mutants lacking these pathways to remain alive for long periods of time and, in some cases, to even reproduce as homozygotes under standard or specially controlled lab conditions.
We started by simply analyzing the function of the Gq pathway separate from the other pathways. Studies from the Kaplan lab and others had suggested that the synaptic vesicle priming protein UNC-13 might be a major downstream target of the DAG produced by the Gq pathway. Could the major function of the Gq pathway be to cause SV release through a DAG and UNC-13-dependent mechanism? To test this, we first asked if could bypass the EGL-30 (Gq) pathway by simply putting animals on plates containing an optimal level of a DAG analog called phorbol myristate acetate (phorbol ester). This is a very hydrophobic small molecule that soaks into the animal and into its neurons quickly.
egl-30 (Gq) null mutants arrest as tiny paralyzed larvae, but they can live for several weeks on culture plates, pumping in a small amount of bacterial food every once in a while due to the myogenic activity of the pharynx, but not growing past the second or third larval stage. This is similar to the phenotype that is seen when animals can't release neurotransmitter. However, that same phenotypes could also be caused by neuronal development defects or muscle defects that have nothing to do with the synaptic function of neurons. However, we were able to show that their paralysis and growth defects resulted from an inability to produce the DAG signal necessary for synaptic transmission and locomotion.
To show this, we simply placed the arrested egl-30 larvae on plates containing various concentrations of phorbol ester, waited 2 hours, and then observed the animals and quantified their locomotion rates. We were able to find a phorbol ester concentration that restored wild type levels of locomotion to egl-30 larvae and that allowed them to escape their larval arrest and grow for several generations on the plates.

To explain these results, we hypothesized that phorbol ester, by mimicking the DAG produced by the Gq pathway, bypassed the requirement for Gq by restoring neurotransmitter release and thus rescuing the paralysis. Since acetylcholine (ACh) is the major excitatory neurotransmitter controlling locomotion rate in C. elegans, we tested this idea by placing phorbol-ester-treated egl-30 nulls on plates containing the acetylcholinesterase inhibitor aldicarb. Aldicarb is an inhibitor of acetycholinesterase, which normally breaks down acetycholine after it is released from synaptic vesicles. Since the secreted ACh that accumulates in the presence of aldicarb is toxic, mutations that decrease or increase the steady-state rate of ACh release confer resistance or hypersensitivity to aldicarb (Adventures>1993-1996). egl-30 mutants are normally strongly resistant to aldicarb due to strongly decreased neurotransmitter release, whereas wild type animals on phorbol ester are strongly hypersensitive to aldicarb due to strongly increased neurotransmitter release. However, we found that egl-30 null mutants on phorbol ester were just as hypersensitive to aldicarb as wild type animals.
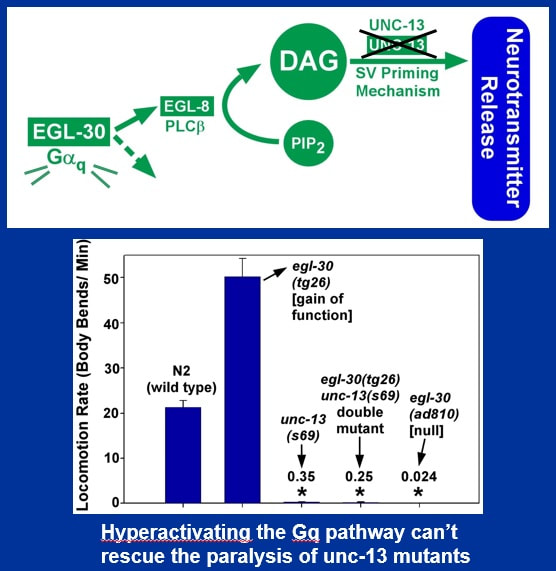
UNC-13, a protein that "primes" synaptic vesicles for the fusion event that releases neurotransmitter, was thought to be a major downstream target of DAG for regulating neurotransmitter release. We found that the action of phorbol ester in stimulating locomotion was completely dependent on UNC-13, since a paralyzed unc-13 null mutant remained paralyzed on phorbol ester.
UNC-13, a protein that "primes" synaptic vesicles for the fusion event that releases neurotransmitter, was thought to be a major downstream target of DAG for regulating neurotransmitter release. We found that the action of phorbol ester in stimulating locomotion was completely dependent on UNC-13, since a paralyzed unc-13 null mutant remained paralyzed on phorbol ester.

Similarly, we found that a mutation that hyperactivates the Gq pathway and causes hyperactive locomotion had no significant effect on the locomotion of unc-13 null mutants. In conclusion the results of our egl-30 pathway epistasis analysis suggested that the core function of the Gq pathway is to promote the neurotransmitter release that drives locomotion (through the production of DAG), and that DAG acts on the neurotransmitter release machinery in an UNC-13-dependent manner.
How does the Gs Pathway affect neurotransmitter release?

The Plasterk lab had produced a nice deletion of the acy-1 (adenylyl cyclase) gene in an earlier study. Although acy-1 nulls are arrested as larvae, they balanced the mutation over a closely linked "Dumpy" mutation so it was extremely easy to maintain as a heterozygote. This was a very important genetic tool for us. acy-1 null mutants are nearly paralyzed, having locomotion rates about 5% of wild type, but not as paralyzed as egl-30 (Gq nulls). In Schade et al., 2005, we used epistasis analysis to show that all of the effects of (GSA-1) Gs on locomotion are channeled through ACY-1 (adenylyl cyclase). Although acy-1 null mutant animals are arrested as larvae, we showed in this study that they can live for over 3 weeks on culture plates.
We were concerned that some of the paralysis might be due to improper development of the nervous system or to muscle defects and not to synaptic function defects. We knew from the Plasterk lab's studies that the Gs pathway was expressed in both body wall muscle and the entire nervous system. We found that we could rescue the larval arrest, but not the paralysis, of acy-1 null mutants by expressing acy-1 in body wall muscle cells. This allowed us to produce adult animals that were null for acy-1 the nervous system. we called these animals "muscle-rescued acy-1 nulls" or "neuronal acy-1 nulls".
We were concerned that some of the paralysis might be due to improper development of the nervous system or to muscle defects and not to synaptic function defects. We knew from the Plasterk lab's studies that the Gs pathway was expressed in both body wall muscle and the entire nervous system. We found that we could rescue the larval arrest, but not the paralysis, of acy-1 null mutants by expressing acy-1 in body wall muscle cells. This allowed us to produce adult animals that were null for acy-1 the nervous system. we called these animals "muscle-rescued acy-1 nulls" or "neuronal acy-1 nulls".

To test whether the paralysis of muscle-rescued acy-1 nulls is caused by defects in the function, versus the development, of the nervous system, we produced a similar transgenic strain that rescued acy-1 in muscle, but also included a second transgene that allowed us to induce expression of the wild type acy-1 cDNA in the nervous system of adult animals using the heat shock promoter. Using this strain we found that, just 2 hours after a 1 hour heat shock, neuronal acy-1 nulls were moving in a coordinated fashion at rates not significantly different from wild type. This suggests that the near-paralysis of neuronal acy-1 nulls is caused by functional, not developmental, defects in neurons.

To test whether the ACY-1 (and thus the Gs pathway) is required for neurotransmitter release, we quantified the sensitivity of acy-1 nulls to aldicarb, which can be incorporated into the culture plates at various concentrations. Whereas mutations that reduce the function of the EGL-30 (Gq) pathway cause strong resistance to aldicarb (indicating impaired release of the neurotransmitter acetylcholine, the muscle-rescued acy-1 nulls had normal wild type sensitivity to aldicarb. This is consistent with our results in Schade et al., 2005 showing that over-expressing an acy-1 gain-of-function mutation in the nervous system does not have a significant effect on aldicarb sensitivity despite causing hyperactive locomotion and suppressing the paralysis of ric-8 reduction-of-function mutants (Adventures 2000-2005A). These results suggest that the neuronal Gs pathway, unlike the Gq pathway, is not required for neurotransmitter release, despite the fact that mutants lacking this pathway are strongly paralyzed due to functional, non-developmental defects in their neurons.

The Relationship of the Gq and Gs Pathways
The Gq and Gs pathways seem to be working together to do two different things to accomplish the same goal: coordinated activation of synapses to cause a behavior, in this case locomotion. Without either pathway animals are paralyzed or near-paralyzed. Each is essential for the behavior, but they appear to be doing different things. To analyze the relationship of these two pathways to each other in living animals, we did epistasis experiments. This is simply a fancy way of saying we made double mutants in which one pathway is completely knocked out and the other is strongly activated and vice versa.
We first asked to what extent the Gs pathway is dependent on the Gq pathway to exert its effects on locomotion. To answer this, we analyzed double mutants containing an activated Gs pathway in combination with an egl-30 (Gq) null mutation. We found that mutations that strongly activated the Gs pathway and normally caused very hyperactive, coordinated locomotion had little or no effect on the paralysis of Gq null mutants. For example, the Gq null was completely unaffected by the kin-2 mutation that leads to hyperactivation of Protein Kinase A. However, this 100% block of activated Protein Kinase A in driving locomotion was not the result of unexpected synthetic effects of combining the two mutations, because placing these double mutant animals on plates containing phorbol ester quickly restored wild type levels of locomotion.
We first asked to what extent the Gs pathway is dependent on the Gq pathway to exert its effects on locomotion. To answer this, we analyzed double mutants containing an activated Gs pathway in combination with an egl-30 (Gq) null mutation. We found that mutations that strongly activated the Gs pathway and normally caused very hyperactive, coordinated locomotion had little or no effect on the paralysis of Gq null mutants. For example, the Gq null was completely unaffected by the kin-2 mutation that leads to hyperactivation of Protein Kinase A. However, this 100% block of activated Protein Kinase A in driving locomotion was not the result of unexpected synthetic effects of combining the two mutations, because placing these double mutant animals on plates containing phorbol ester quickly restored wild type levels of locomotion.

The above results suggest that the ability of the Gs pathway to stimulate locomotion is completely dependent on the Gq pathway, and thus that the Gq and Gs pathway, as distinct as they are in their functions, must converge at some point in their joint regulation of locomotion rate.
We next did the complementary experiment by knocking out the neuronal Gs pathway, via the muscle-rescued acy-1 null, and asking to what extent we could bypass the resulting near-paralysis by adding in a mutation that strongly activates the Gq pathway or by placing the animals on culture plates with phorbol ester. Those results showed that the activated Gq pathway could partially bypass lack of a Gs pathway, restoring locomotion rates to an intermediate level. However, animals spent most of their time (72 +/- 3%) in a tightly "knotted" phenotype, from which they occasionally escaped for short bouts of coordinated locomotion.
We next did the complementary experiment by knocking out the neuronal Gs pathway, via the muscle-rescued acy-1 null, and asking to what extent we could bypass the resulting near-paralysis by adding in a mutation that strongly activates the Gq pathway or by placing the animals on culture plates with phorbol ester. Those results showed that the activated Gq pathway could partially bypass lack of a Gs pathway, restoring locomotion rates to an intermediate level. However, animals spent most of their time (72 +/- 3%) in a tightly "knotted" phenotype, from which they occasionally escaped for short bouts of coordinated locomotion.

The above results suggest that the Gq pathway can still exert its function in stimulating neurotransmitter release independent of the neuronal Gs pathway, but that these conditions do not support stable, coordinated locomotion. Furthermore, because phorbol esters confer the knotted phenotype on mutants lacking a neuronal Gs pathway, these results suggest that the Gs pathway regulates the response to DAG and thus exerts its effects downstream of DAG production. Thus, from a genetic pathway standpoint, the Gs pathway acts on the Gq pathway to convert neurotransmitter release (a function of the Gq pathway) into sustained, coordinated locomotion (a combined function of the Gs and Gq pathways).
The Relationship of RIC-8 to the Gq and Gs Pathways
Our data from Adventures>1996-2000 suggested that RIC-8 was necessary to maintain the Gq pathway in a functional state. However, we didn't yet know if RIC-8 was important to maintain the Gs pathway in a functional state, and Greg Tall's biochemistry had so far demonstrated that RIC-8 was important for Gq and Go function, but he hadn't yet demonstrated a functional effect on Gs.
To further investigate the relationship of RIC-8 to the Gq and Gs pathways, we obtained a ric-8 null deletion mutant from Bob Barstead's C. elegans Gene Knockout lab at OMRF. These animals were paralyzed to the same extent as Gq null mutants. About one third of ric-8 nulls were larval arrested, and two thirds matured to become paralyzed, sterile adults. We were able to rescue all of these phenotypes with the wild type ric-8 gene. We then used the ric-8 null mutant in epistasis analysis, combining it with mutations or drugs that activate the Gq and/ or Gs pathways.
We found that mutations that hyperactivate the Gs pathway were not very effective in rescuing the paralysis of ric-8 null mutants, causing only a tiny, barely significant increase in locomotion rate. Putting ric-8 null mutants on culture plates with phorbol ester caused a more significant increase in locomotion rate, but the locomotion rate of these animals on phorbol ester was still far below the locomotion rate of Gq nulls on phorbol ester. Recall that phorbol ester is a DAG analog that bypasses the Gq pathway because one of the main functions of the Gq pathway is to make DAG. This suggests that ric-8 nulls cannot simply be defective in their Gq pathway. However, when we combined phorbol ester and an activated Gs pathway with the ric-8 null mutation, those animals had their locomotion rates restored to levels slightly greater than that of wild type on the phorbol ester-free control plates (similar to a Gq null on phorbol ester) and, remarkably, showed beautifully coordinated locomotion for the next 30 - 60 minutes.
To further investigate the relationship of RIC-8 to the Gq and Gs pathways, we obtained a ric-8 null deletion mutant from Bob Barstead's C. elegans Gene Knockout lab at OMRF. These animals were paralyzed to the same extent as Gq null mutants. About one third of ric-8 nulls were larval arrested, and two thirds matured to become paralyzed, sterile adults. We were able to rescue all of these phenotypes with the wild type ric-8 gene. We then used the ric-8 null mutant in epistasis analysis, combining it with mutations or drugs that activate the Gq and/ or Gs pathways.
We found that mutations that hyperactivate the Gs pathway were not very effective in rescuing the paralysis of ric-8 null mutants, causing only a tiny, barely significant increase in locomotion rate. Putting ric-8 null mutants on culture plates with phorbol ester caused a more significant increase in locomotion rate, but the locomotion rate of these animals on phorbol ester was still far below the locomotion rate of Gq nulls on phorbol ester. Recall that phorbol ester is a DAG analog that bypasses the Gq pathway because one of the main functions of the Gq pathway is to make DAG. This suggests that ric-8 nulls cannot simply be defective in their Gq pathway. However, when we combined phorbol ester and an activated Gs pathway with the ric-8 null mutation, those animals had their locomotion rates restored to levels slightly greater than that of wild type on the phorbol ester-free control plates (similar to a Gq null on phorbol ester) and, remarkably, showed beautifully coordinated locomotion for the next 30 - 60 minutes.

Since both the Gq and Gs pathways had to be hyperactivated and/ or bypassed with phorbol ester to rescue the ric-8 null mutant, the above results suggest that RIC-8 is required to maintain both the Gq and the Gs pathways in a functional state.
An Updated Pathway Model
As a result of our epistasis experiments, we updated our pathway model, calling it the Synaptic Signaling Network to reflect the interaction of three major G protein pathways in bridging the gap between synaptic function and behavior. Many questions remained unanswered, as indicated by the dashed lines, gaps, and empty rectangles. Some of these questions were answered or further addressed in later studies. However, the updated model as of this study reflects our findings that the Gs pathway is dependent on the Gq pathway to exert its effects on the locomotion behavior, that the Gs pathway is involved in the locomotion response to the DAG produced by the Gq pathway (i.e. that the Gs pathway interacts with the Gq pathway downstream of DAG), and that RIC-8 is required to maintain both the Gq and the Gs pathways in a functional state.

Reynolds NK, Schade MA, and Miller KG (2005). Convergent, RIC-8 – dependent Ga signaling pathways in the Caenorhabditis elegans synaptic signaling network. Genetics. 169(2), 651-670. PMCID: PMC1449085.
Published back-to-back with Schade et al., 2005.
Cited by 57 papers as of April, 2018.
Published back-to-back with Schade et al., 2005.
Cited by 57 papers as of April, 2018.
Remaining Important Questions...and some Comments
-While the function of the Gq pathway as a neurotransmiter release regulator is somewhat clear, the presice function of the Gs pathway remains much less clear. How could the neuronal Gs pathway be so critical for the locomotion behavior without affecting muscle function or the development of neurons or synapses, and without affecting overall levels of acetylcholine release in the C. elegans nervous system? One possibility that we proposed in our paper is that the Gs pathway helps determine the specific synapses that are optimal for locomotion, leading to those synapses being turned ON or “activated”. Once turned ON, an optimal synapse would release more neurotransmitter, but this wouldn’t be detected in the whole animal aldicarb assay, which sums all of the thousands of motor neuron synapses. The optimal synapses would simply be lost in the noise of the other synapses. According to this model, in a mutant lacking a neuronal Gs pathway, the nervous system can no longer identify which synapses are optimal for locomotion. Neurotransmitter release, driven by the Gq pathway, still occurs at near-normal levels, but it is uncoupled from locomotion, thus leading to the near paralysis of animals lacking a neuronal Gs pathway. Forcing global neurotransmitter release in this context by hyperactivating the Gq pathway or soaking in phorbol ester results in a “knotted” phenotype as release from non-optimal synapses erases the effects of release from optimal synapses.
-Although our study predicts that the Gs and Gq pathways interact downstream of DAG, our results provide no clues about the molecular targets that allow the Gs pathway to transduce behaviorally relevant signals onto the Gq pathway. Instead our study broadly examined the relationship between the pathways from a whole animal, behavioral and neurotransmitter release perspective.
-Our analysis of double mutants containing an activated Gq pathway, or animals on phorbol ester, in combination with a near-null unc-13 mutation suggest that all effectors of the presynaptic Gq pathway (including DAG) ultimately channel their effects through UNC-13 or an UNC-13-dependent process. However, in light of the findings by Rhee et al., 2002, who showed that eliminating the ability of UNC-13 to bind DAG does not significantly affect neurotransmitter release, we believe that much of this requirement must be indirect, such as through Protein Kinase C phosphorylating UNC-13 or UNC-13-interacting proteins. PKC is a known DAG binding protein, and there are several PKC isoforms in C. elegans that need to be knocked out, combined together, and analyzed. We and others have found that the PKC isoform TPA-1 is required for the hyperactive locomotion response induced by phorbol ester.
-Although our study predicts that the Gs and Gq pathways interact downstream of DAG, our results provide no clues about the molecular targets that allow the Gs pathway to transduce behaviorally relevant signals onto the Gq pathway. Instead our study broadly examined the relationship between the pathways from a whole animal, behavioral and neurotransmitter release perspective.
-Our analysis of double mutants containing an activated Gq pathway, or animals on phorbol ester, in combination with a near-null unc-13 mutation suggest that all effectors of the presynaptic Gq pathway (including DAG) ultimately channel their effects through UNC-13 or an UNC-13-dependent process. However, in light of the findings by Rhee et al., 2002, who showed that eliminating the ability of UNC-13 to bind DAG does not significantly affect neurotransmitter release, we believe that much of this requirement must be indirect, such as through Protein Kinase C phosphorylating UNC-13 or UNC-13-interacting proteins. PKC is a known DAG binding protein, and there are several PKC isoforms in C. elegans that need to be knocked out, combined together, and analyzed. We and others have found that the PKC isoform TPA-1 is required for the hyperactive locomotion response induced by phorbol ester.
Fun Factoids about this Study
-We find it extremely interesting that treating Gq null mutants in a non-localized manner with an optimal concentration of a DAG analog (phorbol ester) rescues their paralysis to wild type levels of coordinated locomotion! Recall that we just put these paralyzed animals on plates with phorbol ester which, over the next 2 hours, soaked into the animals’ nervous systems. This, and other related findings, suggest that DAG is a critical component for synaptic transmission in the context of intact behaving animals.
-Our study was the first to investigate the relationship of the Gq and Gs pathways to each other in intact behaving animals.
-Our study was published back-to-back with Schade et al., 2005 (Adventures>2000-2005A).
-Our study was the first to investigate the relationship of the Gq and Gs pathways to each other in intact behaving animals.
-Our study was published back-to-back with Schade et al., 2005 (Adventures>2000-2005A).
Related Studies
We followed up on this study with three major publications. Charlie et al., 2006A "completes" the Gs pathway by showing that the two large genetic screens in this study also netted a single rare mutation in pde-4, which encodes the cAMP Phosphodiesterase that is part of the Gs pathway and an ortholog of the Drosophila "Dunce" learning and memory mutant (Adventures > 2004-2006). Charlie et al., 2006B further investigates the relationship of the Gs and Gq pathways to each other and to UNC-31 (CAPS), which, among other possible functions, is involved in the release of neuropeptides by dense core vesicles (Adventures>2006). Finally, Williams et al., 2007 reports our discovery of the missing effector branch of the Gq pathway (Adventures>2005-2007).
Reynolds NK, Schade MA, and Miller KG (2005). Convergent, RIC-8 – dependent G alpha signaling pathways in the Caenorhabditis elegans synaptic signaling network. Genetics. 169(2), 651-670. PMCID: PMC1449085.
Charlie NK, Thomure AM, Schade MA, and Miller KG (2006). The Dunce cAMP phosphodiesterase PDE-4 negatively regulates Gs – dependent and Gs – independent cAMP pools in the Caenorhabditis Elegans synaptic signaling network. Genetics 173(1), 111-130. PMCID: PMC1461419.
Williams SL, Lutz S, Charlie NK, Vettel C, Ailion M, Coco C, Tesmer JJ, Jorgensen EM, Wieland T, and Miller KG (2007). Trio’s Rho – Specific GEF domain is the missing Gq Effector in C. elegans. Genes Dev. 21:2731-2746. PMCID: PMC2045128.
The Gs pathway was also studied genetically in the fruit fly Drosophila melanogaster, although largely not in intact behaving animals. Electrophysiological studies of Drosophila Gs null and reduction-of-function mutants also found normal or slightly increased neurotransmitter release in response low frequency artificial nerve stimulation. These studies also found that Gs mutants, unlike wild type, showed no potentiation (increase) in neurotransmitter release when stimulated again after a series of high frequency stimulations. Like worm mutants lacking a neuronal Gs pathway, fly larvae lacking Gs show little or no movement. The strong contrast between the behavioral and physiological effects of decreased cAMP is puzzling and suggests that we do not adequately understand what cAMP is doing at the synapse.
Wolfgang, W.J., Hoskote, A., Roberts, I.J., Jackson, S. and Forte, M., 2001 Genetic analysis of the Drosophila Gs gene. Genetics 158: 1189-1201.
Hou, D., K. Suzuki, W. J. Wolfgang, C. Clay, M. Forte et al., 2003 Presynaptic impairment of synaptic transmission in Drosophila embryos lacking Gs. J Neurosci 23: 5897-5905.
Renden, R. B., and K. Broadie, 2003 Mutation and Activation of Gs Similarly Alters Pre- and Postsynaptic Mechanisms Modulating Neurotransmission. J. Neurophysiol. 89: 2620-2638.
Although our study was the first to investigate the relationship of the Gq and Gs pathways to each other in intact behaving animals, previous studies in cultured primary neurons from Aplysia and Crayfish seem consistent with the possibility that co-activation of the Gq and Gs pathways mediates synapse activation/ turns synapses ON. These studies, from the labs of Mark Klein and Robert Zucker, showed that application of the neurotransmitter serotonin, which can activate both the Gq and Gs pathways in Aplysia sensory neurons, induces rapid synapse “maturation”. The resulting synaptic facilitation (increase in release) occurs by means of an “all-or-none” switching ON of synapses. A study of serotonin’s effects at the crayfish neuromuscular junction proposed a similar conclusion as a possibility.
Coulson, R. L., and M. Klein, 1997 Rapid development of synaptic connections and plasticity between sensory neurons and motor neurons of Aplysia in cell culture: implications for learning and regulation of synaptic strength. J Neurophysiol 77: 2316-2327.
Royer, S., R. L. Coulson and M. Klein, 2000 Switching off and on of synaptic sites at aplysia sensorimotor synapses. J Neurosci 20: 626-638.
Wang, C., and R. S. Zucker, 1998 Regulation of synaptic vesicle recycling by calcium and serotonin. Neuron 21: 155-167.
In other intriguing studies, in which the activity state of the Gq pathway was unknown or not controlled, manipulations that increased cAMP in primary cultures of vertebrate neurons “activated” previously “silent” synapses.
Tong, G., R. C. Malenka and R. A. Nicoll, 1996 Long-term potentiation in cultures of single hippocampal granule cells: a presynaptic form of plasticity. Neuron 16: 1147-1157.
Chavis, P., P. Mollard, J. Bockaert and O. Manzoni, 1998 Visualization of cyclic AMP-regulated presynaptic activity at cerebellar granule cells. Neuron 20: 773-781.
Ma, L., L. Zablow, E. R. Kandel and S. A. Siegelbaum, 1999 Cyclic AMP induces functional presynaptic boutons in hippocampal CA3-CA1 neuronal cultures. Nature Neurosci. 2: 24-30.
In 2003, before this publication, Greg Tall demonstrated that RIC-8 has guanine nucleotide exchange (GEF) activity for Gq and Go, but he was unable to demonstrate that for Gs at that time. Our prediction that RIC-8 is required for Gs signalling was proven true a few years later when Greg Tall and another lab demonstrated GEF activity of frog and mammalian RIC-8B for Gs.
Tall, GG, Krumins AM and AG Gilman (2003). Mammalian Ric-8A (Synembryn) Is a Heterotrimeric G alpha Protein Guanine Nucleotide Exchange Factor. J Biol Chem 278: 8356-8362.
Chan, P, Gabay M, Wright, FA, Tall, G.G. (2011). RIC-8B is a GTP-dependent G Protein alpha S Guanine Nucleotide Exchange Factor. J Biol Chem 286: 19932-42.
Ximena, R, Pasten P, MartInez, S, et al. (2008) xRIC-8 is a GEF for Gs and participates in maintaining meiotic arrest in Xenopus laevis oocytes. J Cell Physiol 214: 673-80.
The Plasterk lab produced null mutant in acy-1 that was so critical for our study and nicely balanced it with a Dpy mutation.
Moorman, C., and R. H. Plasterk, 2002 Functional characterization of the adenylyl cyclase gene sgs-1 by analysis of a mutational spectrum in Caenorhabditis elegans. Genetics 161: 133-142.
Reynolds NK, Schade MA, and Miller KG (2005). Convergent, RIC-8 – dependent G alpha signaling pathways in the Caenorhabditis elegans synaptic signaling network. Genetics. 169(2), 651-670. PMCID: PMC1449085.
Charlie NK, Thomure AM, Schade MA, and Miller KG (2006). The Dunce cAMP phosphodiesterase PDE-4 negatively regulates Gs – dependent and Gs – independent cAMP pools in the Caenorhabditis Elegans synaptic signaling network. Genetics 173(1), 111-130. PMCID: PMC1461419.
Williams SL, Lutz S, Charlie NK, Vettel C, Ailion M, Coco C, Tesmer JJ, Jorgensen EM, Wieland T, and Miller KG (2007). Trio’s Rho – Specific GEF domain is the missing Gq Effector in C. elegans. Genes Dev. 21:2731-2746. PMCID: PMC2045128.
The Gs pathway was also studied genetically in the fruit fly Drosophila melanogaster, although largely not in intact behaving animals. Electrophysiological studies of Drosophila Gs null and reduction-of-function mutants also found normal or slightly increased neurotransmitter release in response low frequency artificial nerve stimulation. These studies also found that Gs mutants, unlike wild type, showed no potentiation (increase) in neurotransmitter release when stimulated again after a series of high frequency stimulations. Like worm mutants lacking a neuronal Gs pathway, fly larvae lacking Gs show little or no movement. The strong contrast between the behavioral and physiological effects of decreased cAMP is puzzling and suggests that we do not adequately understand what cAMP is doing at the synapse.
Wolfgang, W.J., Hoskote, A., Roberts, I.J., Jackson, S. and Forte, M., 2001 Genetic analysis of the Drosophila Gs gene. Genetics 158: 1189-1201.
Hou, D., K. Suzuki, W. J. Wolfgang, C. Clay, M. Forte et al., 2003 Presynaptic impairment of synaptic transmission in Drosophila embryos lacking Gs. J Neurosci 23: 5897-5905.
Renden, R. B., and K. Broadie, 2003 Mutation and Activation of Gs Similarly Alters Pre- and Postsynaptic Mechanisms Modulating Neurotransmission. J. Neurophysiol. 89: 2620-2638.
Although our study was the first to investigate the relationship of the Gq and Gs pathways to each other in intact behaving animals, previous studies in cultured primary neurons from Aplysia and Crayfish seem consistent with the possibility that co-activation of the Gq and Gs pathways mediates synapse activation/ turns synapses ON. These studies, from the labs of Mark Klein and Robert Zucker, showed that application of the neurotransmitter serotonin, which can activate both the Gq and Gs pathways in Aplysia sensory neurons, induces rapid synapse “maturation”. The resulting synaptic facilitation (increase in release) occurs by means of an “all-or-none” switching ON of synapses. A study of serotonin’s effects at the crayfish neuromuscular junction proposed a similar conclusion as a possibility.
Coulson, R. L., and M. Klein, 1997 Rapid development of synaptic connections and plasticity between sensory neurons and motor neurons of Aplysia in cell culture: implications for learning and regulation of synaptic strength. J Neurophysiol 77: 2316-2327.
Royer, S., R. L. Coulson and M. Klein, 2000 Switching off and on of synaptic sites at aplysia sensorimotor synapses. J Neurosci 20: 626-638.
Wang, C., and R. S. Zucker, 1998 Regulation of synaptic vesicle recycling by calcium and serotonin. Neuron 21: 155-167.
In other intriguing studies, in which the activity state of the Gq pathway was unknown or not controlled, manipulations that increased cAMP in primary cultures of vertebrate neurons “activated” previously “silent” synapses.
Tong, G., R. C. Malenka and R. A. Nicoll, 1996 Long-term potentiation in cultures of single hippocampal granule cells: a presynaptic form of plasticity. Neuron 16: 1147-1157.
Chavis, P., P. Mollard, J. Bockaert and O. Manzoni, 1998 Visualization of cyclic AMP-regulated presynaptic activity at cerebellar granule cells. Neuron 20: 773-781.
Ma, L., L. Zablow, E. R. Kandel and S. A. Siegelbaum, 1999 Cyclic AMP induces functional presynaptic boutons in hippocampal CA3-CA1 neuronal cultures. Nature Neurosci. 2: 24-30.
In 2003, before this publication, Greg Tall demonstrated that RIC-8 has guanine nucleotide exchange (GEF) activity for Gq and Go, but he was unable to demonstrate that for Gs at that time. Our prediction that RIC-8 is required for Gs signalling was proven true a few years later when Greg Tall and another lab demonstrated GEF activity of frog and mammalian RIC-8B for Gs.
Tall, GG, Krumins AM and AG Gilman (2003). Mammalian Ric-8A (Synembryn) Is a Heterotrimeric G alpha Protein Guanine Nucleotide Exchange Factor. J Biol Chem 278: 8356-8362.
Chan, P, Gabay M, Wright, FA, Tall, G.G. (2011). RIC-8B is a GTP-dependent G Protein alpha S Guanine Nucleotide Exchange Factor. J Biol Chem 286: 19932-42.
Ximena, R, Pasten P, MartInez, S, et al. (2008) xRIC-8 is a GEF for Gs and participates in maintaining meiotic arrest in Xenopus laevis oocytes. J Cell Physiol 214: 673-80.
The Plasterk lab produced null mutant in acy-1 that was so critical for our study and nicely balanced it with a Dpy mutation.
Moorman, C., and R. H. Plasterk, 2002 Functional characterization of the adenylyl cyclase gene sgs-1 by analysis of a mutational spectrum in Caenorhabditis elegans. Genetics 161: 133-142.
