2009-2014: A novel pathway that inhibits the release of neuropeptides from neuronal cell somas
Neurons use Dense Core Vesicles (DCVs) to release neuropeptides. Neuropeptides complement the classical small molecule neurotransmitters that are released by Synaptic Vesicles (SVs). Neuropeptides are kind of like the brain's hormones since they can act over long or short distances to activate entire circuits of neurons to turn synapses ON or OFF and thus cause or prevent the release of small molecule neurotransmitters via SVs.
In fact, some of the hormones released by the endocrine system are neuropeptides that are released via DCVs. In the endocrine system DCVs often release neuropeptides directly from cell somas, whereas in true neurons most DCVs are shipped to axons where they are released from synapses.
In this Adventure we discovered a pathway that prevents DCVs from releasing neuropeptides in neuronal cell somas, thus allowing DCVs to ship their neuropeptide cargo to axons for storage and release upon receiving the appropriate signal.
One big surprise of our study is that the pathway relies on a protein called CaM Kinase II (UNC-43 in C. elegans). The reason this was a surprise is because CaM Kinase II was discovered biochemically 34 years before our paper was published, and it appeared as the main topic of hundreds of prominant publications. However, although it was known to have several important functions in the brain and to make up 1-2% of total brain protein, it was not known to have the critical DCV function that we discovered.
Our study thus emphasizes and highlights the power of unbiased forward genetic screens in model organisms for uncovering new functions, even for heavily investigated proteins.
As with most "forward genetic" studies in model organisms, we didn't begin our study looking for a new function for UNC-43 (CaM Kinase II). Instead, we used a forward genetic screen to ask the organism (C. elegans) to help us find all of the proteins that are important for neurons to make DCVs and/ or to ship the DCVs from cell somas to axons (see Genetic Screens Explained).
In this genetic screen, we used a genomically integrated transgene to add a fluoresent protein to the outside surface of DCVs in a small number of neurons in the ventral nerve cord of the animal. The DCVs we labeled are in neurons that have their cell somas on the ventral side of the animal and their axons with synapses on the dorsal side of the animal. The somas and the axons are connected by commisures, which are thin tubes that extend from the soma. DCVs and SVs travel through commissures to reach synapses in the axons.
In our genetic screen, we simply mutagenized animals and screened their grandprogeny for mutants in which the distribution of DCVs between somas and axons was obviously abnormal.
In fact, some of the hormones released by the endocrine system are neuropeptides that are released via DCVs. In the endocrine system DCVs often release neuropeptides directly from cell somas, whereas in true neurons most DCVs are shipped to axons where they are released from synapses.
In this Adventure we discovered a pathway that prevents DCVs from releasing neuropeptides in neuronal cell somas, thus allowing DCVs to ship their neuropeptide cargo to axons for storage and release upon receiving the appropriate signal.
One big surprise of our study is that the pathway relies on a protein called CaM Kinase II (UNC-43 in C. elegans). The reason this was a surprise is because CaM Kinase II was discovered biochemically 34 years before our paper was published, and it appeared as the main topic of hundreds of prominant publications. However, although it was known to have several important functions in the brain and to make up 1-2% of total brain protein, it was not known to have the critical DCV function that we discovered.
Our study thus emphasizes and highlights the power of unbiased forward genetic screens in model organisms for uncovering new functions, even for heavily investigated proteins.
As with most "forward genetic" studies in model organisms, we didn't begin our study looking for a new function for UNC-43 (CaM Kinase II). Instead, we used a forward genetic screen to ask the organism (C. elegans) to help us find all of the proteins that are important for neurons to make DCVs and/ or to ship the DCVs from cell somas to axons (see Genetic Screens Explained).
In this genetic screen, we used a genomically integrated transgene to add a fluoresent protein to the outside surface of DCVs in a small number of neurons in the ventral nerve cord of the animal. The DCVs we labeled are in neurons that have their cell somas on the ventral side of the animal and their axons with synapses on the dorsal side of the animal. The somas and the axons are connected by commisures, which are thin tubes that extend from the soma. DCVs and SVs travel through commissures to reach synapses in the axons.
In our genetic screen, we simply mutagenized animals and screened their grandprogeny for mutants in which the distribution of DCVs between somas and axons was obviously abnormal.

After isolating the mutants we then mapped the mutations to specific genes. Since each gene encodes a protein that the animal uses, this allowed us to identify the proteins that are important for ensuring that DCVs accumulate at optimal levels in the synaptic regions of axons. Two of the mutations we isolated mapped to a gene known as unc-43. UNC-43 encodes the C. elegans ortholog of Calcium-Calmodulin Kinase two, or CaM Kinase II for short. A kinase is a protein that phosphorylates (adds a phosphate group) to another protein, which usually activates or inhibits the target protein. Phosphorylation is one of the most common ways to send signals between proteins to turn proteins/ enzymes ON or OFF. Both mutations cause early STOP codons in the kinase domain and thus are predicted to reduce or eliminate the function of the protein.

|
The green fluorescent protein (GFP) that we used to "see" DCVs in living animals was attached to a protein that was embedded in the membrane of the DCV. Quantitative imaging revealed that only about 20-30% of that tagged protein made it to axons. However, at that point we didn't know if that meant that fewer DCVs were reaching axons or if that protein was getting inappropriately stripped off the the DCVs during DCV biogenesis. The last images/ bars show that we can rescue this phenotype by expressing the wild type unc-43 cDNA in the same neurons but that an unc-43 cDNA in which the kinase domain has been mutated does not rescue. This latter result shows that the kinase activity of CaM Kinase II is required for this function as opposed to some other domain of the protein.
Quantitative imaging revealed that both unc-43 mutants had wild type levels of the the fluorescent DCV membrane cargo cargo in their somas. One problem with the fluorescent membrane marker is that it doesn't tell you if the fluorescence is associated with intact DCVs versus other structures such as the plasma membrane. For example, if a DCV fused with the plasma membrane to release its neuropeptide cargo, its membrane would now be incorporated into the plasma membrane, and that fluorescently tagged protein could "hang around" a long time in the plasma membrane. Because of this problem, we wanted to look at other cargos that more faithfully coincide with actual DCVs. |


|
In a previous study, we had shown that neuropeptide cargo, which is often aggregated into the "dense core" of the DCV faithfully represents actual DCVs. There are 2 ways to "tag" aggregated neuropeptide cargo with a fluorescent protein. One is to attach the fluorescent tag (in this case "Venus") to a neuropeptide that is not processed/ cut into smaller neuropeptides, such as the INS-22 peptide. That way the fluorescent tag is not cut away from the aggregated neuropeptides during DCV maturation. The second way is to attach the tag to a neuropeptide that is normally processed/ cut into smaller pieces but to transfer the transgene with the tagged peptide into a genetic background that lacks the processing enzyme that normally cuts the neuropeptide into smaller pieces.
We used both of the above methods to tag neuropeptide cargo and found that about 90% of the neuropeptide cargo was missing from the axons of mutants lacking UNC-43 (CaM Kinase II).
We used both of the above methods to tag neuropeptide cargo and found that about 90% of the neuropeptide cargo was missing from the axons of mutants lacking UNC-43 (CaM Kinase II).

|
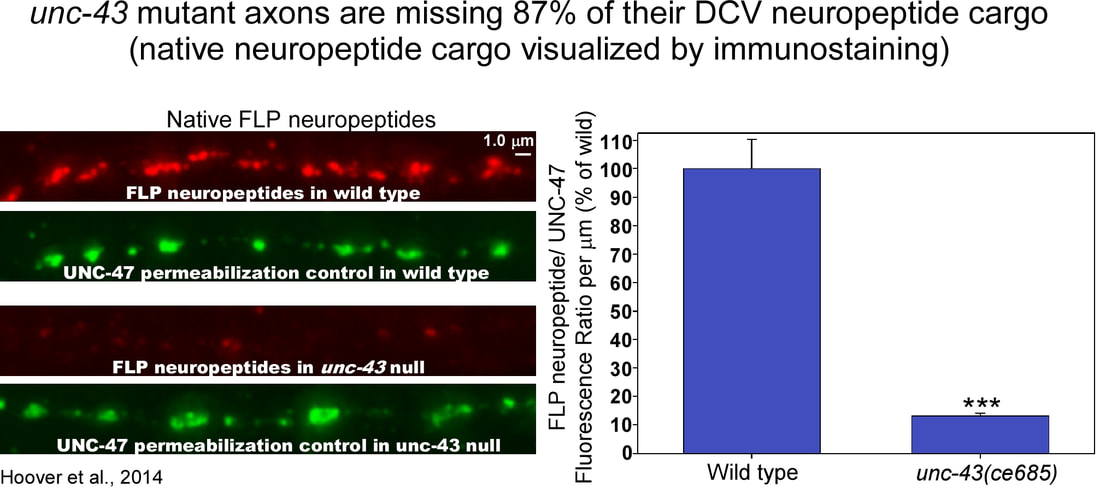
We got similar results when we imaged native neuropeptide cargos called FLP neuropeptides using quantitative immunstaining with an antibody that recognizes FLP neuropeptides. An unc-43 null mutant was missing 87% of this native neuropeptide cargo in its axons. However, when we imaged a native synaptic vesicle cargo using quantitative immunostaining, its levels were only slightly reduced in unc-43 (CaM Kinase II) mutant axons. Thus, UNC-43 appears to be less important for SVs than for DCVs. To determine whether the DCV cargo losses in unc--43 mutant axons correspond to similar reductions in actual DCV numbers, we recruited Dr. Janet Richmond and one of her graduate students to use the technique of high-pressure freezing electron microscopy to count DCVs in unc-43 mutant synapses. These counts were done blinded to genotype. The two unc-43 mutants had 78% and 94% fewer DCVs at their synapses than wild type animals. In contrast SV numbers were only reduced by 7% (not significant) and 24% (significant) in the two mutants. Thus, the EM data support the quantitative fluorescence data and show that the missing DCV cargos in unc-43 mutant axons correspond to similar reductions in actual DCVs. "CaM Kinase" stands for "Calcium-Calmodulin-activated Kinase". What is the source of the calcium that activates the kinase? To address this, we analyzed mutants in which calcium entry is inhibited from various sources in neurons. Neurons often use calcium as a signal. For some functions the calcium enters from outside the neuron through channels in the plasma membrane. For other functions, the calcium is released from internal stores in endoplasmic reticulum (internal stores). When we blocked or reduced extracellular calcium entry we didn't see any effect on neuropeptide accumulation in axons. However, when we blocked calcium entry from internal stores, we saw a significant reduction in neuropeptide accumulation in axons, although not as strongly reduced as in unc-43 mutants. This suggests that one source of the calcium that activates CaM Kinase II's DCV function may be the ryanodine channel that releases calcium from internal stores in the endoplasmic reticulum. The large reduction of DCVs in unc-43 mutant axons could result either from too much exocytosis of DCVs upon reaching the synaptic region of axons or from a failure of DCVs to exit the cell soma and enter axons. To distinguish between these two possibilities, we counted DCVs in commissures. A commissure is the initial segment of the axon that does not contain synapses. It contains only those DCVs that the neuron is transporting between the cell soma and the distal synaptic region of the axon. In unc-43 mutants, we found that the number of DCVs, tagged on soluble cargo or aggregated neuropeptide cargo, was reduced by 63% and 82%, respectively. These results thus suggest that most DCVs do not properly exit the cell somas of unc-43 mutants or that they exit the cell soma but are secreted from the axon initial segment before being transported to the synaptic region. The above results suggest that fewer DCVs are being transported out the soma into axons in unc-43 mutants. If this is the case then maybe the main DCV-related function of UNC-43 (CaM Kinase II) is to transport DCVs out of the soma into axons. If this is true then we would predict a buildup of DCVs in unc-43 mutant somas, and that buildup may even spill over to dendrites. However, instead of finding a buildup of DCVs in cell somas and dendrites, we found significantly fewer DCVs in unc-43 somas and dendrites compared to wild type. This led us to hypothesize that maybe unc-43 mutants are inappropriately exocytosing their DCVs from cell somas instead shipping most of them to axons before undergoing regulated secretion of DCVs. To determine the extent to which the missing neuropeptide cargos are secreted, we looked for the fluorescently-tagged cargos coelomocytes. Coelomocytes are scavenger cells in the animal's body that endocytose fluid from the body cavity (called the pseudocoelom) and concentrate secreted proteins in their endosomes. The Kaplan lab ingenuously developed this as an assay for quantifying INS-22-Venus neuropeptide secretion in living animals. Using this assay, we found that, even though unc-43 null mutants have fewer DCVs in their soma and axons, they secrete just as much as wild type. This suggests that a substantial amount of the neuropeptide cargo that fails to enter unc-43 mutant axons via DCVs is secreted prior to axonal transport. According to this hypothesis, the neuropeptide cargo in wild type coelomocytes originates mostly from axonal release from DCVs, whereas the neuropeptide cargo in unc-43 mutant coelomocytes originates mostly from cell soma and/ or dendritic release, resulting in the observed wild type neuropeptide levels in unc-43 mutant coelomocytes. To test whether the neuropeptide secretion in unc-43 mutants is regulated (occurring only in response to appropriate signals) or constitutive (occuring immediately and constantly), we used the coelomocyte uptake assay to test whether the neuropeptide secretion requires UNC-31 (CAPS), which is a protein that docks/ primes DCVs for regulated secretion. Using this assay, we found that an unc-31 null mutant accumulated neuropeptide in its coelomocytes at about 50% of wild type, consistent with the partial block in neuropeptide secretion observed in prior studies. However, in unc-43 unc-31 double null mutants, secreted neuropeptide in coelomocytes was reduced by 96% relative to unc-43 single mutants. This suggests that most of the neuropeptide that is secreted from unc-43 mutant neurons exits via an UNC-31-dependent regulated secretory pathway rather than constitutive secretion. The transgenic array that we used for this experiment also expresses a soluble, constitutively secreted form of the fluorescent protein "mCherry" from the same neurons. By imaging the same coelomocytes from mCherry fluorscence, we found that unc-43 mutants have wild type levels of constitutive secretion. Constitutive secretion was not decreased in unc-43; unc-31 double mutants was not decreased and was in fact significantly increased compared to wild type. This may be an indirect result of the near complete block in regulated secretion. |









|
Taken together, the above data suggest that, in the absence of UNC-43 (CaM Kinase II), DCVs are secreted prior to axonal transport via an UNC-31-dependent regulated secretory pathway. If this is true, then blocking DCV secretion using unc-43 unc-31 double mutants should restore wild type or near-wild-type levels of DCVs to axons by allowing them to escape the cell soma instead of being exocytosed. To test this, we quantified neuropeptide cargo in the axons of the same strains we used for the above coelomocyte uptake assay. The results showed that unc-43 unc-31 double mutants accumulate neuropeptide cargo in axons at levels that are not significantly different from unc-31 single mutants and that are about 70% of wild type. Thus, blocking regulated secretion in somas with an unc-31 mutation allows most DCVs in unc-43 mutants to be successfully transported to axons.

|
Although the above data suggest that many DCVs are secreted prior to axonal transport via a regulated secretory pathway, that model does not preclude an additional defect in the active transport of DCVs from cell somas to axons in unc-43 mutants, since this phenotype would be masked if DCVs are lost by secretion prior to transport.
To determine whether loss of UNC-43 affects axonal transport of DCVs in intact animals, we used time-lapse microscopy to analyze DCV movements in the axonal commissures of mutants lacking UNC-43. Since most DCVs fail to enter the axonal commissure in unc-43 single mutants, we analyzed DCV movements in unc-43 unc-31 double mutants and compared them to unc-31 single mutants and wild type. The images on the left show representative kymographs for each strain. A kymograph traces all of the paths/ tracks of DCVs in a defined region over a defined time period. The fastest movements are lines that are closest to horizontal. Our paper shows quantitative data for 5 different movement parameters. The bottom line is that unc-43 mutants showed no significant defect in the axonal tranport of DCVs. |

|
The above data suggest that wild type neurons have a mechanism to inhibit the secretion of neuropeptides from DCVs in the soma. Inhibiting release would allow the KIF1A motor to transport DCVs out of cell somas to the synaptic region of axons where they could be stored until appropriate signals stimulate their release.
If this is true, then wild type animals should have relatively low concentrations of DCVs in their somas and much higher concentrations in axons. To test this prediction, we recruited Maike Kittelmann in Stefan Eimer's lab to reconstruct two motor neuron cell somas along with several motor neuron synapses at high resolution using serial electron micrographs and then counted the DCVs in each region.
Maike found that each soma had about 40 DCVs, which was about 2% of the concentration of DCVs in axons. Thus, although there are no data to rule out that secretion of DCVs normally occurs from wild type motor neuron somas, the reconstruction data suggest that motor neurons do not use somas to store a large resevoir of DCVs for secretion. This contrasts with neuroendocrine cells, such as chromaffin cells and pancreatic beta-cells, in which DCVs number in the tens of thousands per cell.
If this is true, then wild type animals should have relatively low concentrations of DCVs in their somas and much higher concentrations in axons. To test this prediction, we recruited Maike Kittelmann in Stefan Eimer's lab to reconstruct two motor neuron cell somas along with several motor neuron synapses at high resolution using serial electron micrographs and then counted the DCVs in each region.
Maike found that each soma had about 40 DCVs, which was about 2% of the concentration of DCVs in axons. Thus, although there are no data to rule out that secretion of DCVs normally occurs from wild type motor neuron somas, the reconstruction data suggest that motor neurons do not use somas to store a large resevoir of DCVs for secretion. This contrasts with neuroendocrine cells, such as chromaffin cells and pancreatic beta-cells, in which DCVs number in the tens of thousands per cell.

|
If unc-43 mutants secrete native neuropeptides abnormally from their motor neuron axons, this could affect the animal's locomotion behavior. The 90% reduction in DCVs in motor neuron axons could also affect locomotion. To determine the extent to which the sluggish locomotion of unc-43 mutants is caused by improperly acting or missing neuropeptides, versus other functions of UNC-43 that might impact locomotion, we compared the basal locomotion rates of unc-43 mutants in the presence and absence of native neuropeptides. egl-3 null mutants lack functional neuropeptides for locomotion due to a defect in a processing enzyme, and their locomotion rate is about 25% of wild type. unc-43 null mutants have a locomotion rate that is about 60% of wild type. unc-43; egl-3 double mutants have locomotion rates not significantly different from egl-3 null mutants. If unc-43 had other major functions in locomotion that were unrelated to neuropeptides then the unc-43; egl-3 double should be worse then the egl-3 single. Instead our data suggest that the functions of UNC-43 (CaM Kinase II) in locomotion largely overlap with neuropeptide function. Since unc-43 null mutants have locomotion rates that are less impaired than egl-3 mutants, our data also suggest that neuropeptide function is only partially disrupted in an unc-43 null mutant.
|

|
Hoover CM, Edwards SL, Yu S, Kittelmann M, Richmond JE, Eimer S, Yorks RM, and Miller KG. (2014). A Novel CaM Kinase II Pathway Controls the Location of Neuropeptide Release from Caenorhabditis elegans Motor Neurons. Genetics 196(3): 745-765*. PMCID: PMC3948804**
*Featured article. See journal commissioned Commentary in same issue: 196(3): 601-601 in same issue.
**Highlighted in 2014 Genetics Spotlight booklet.
*Featured article. See journal commissioned Commentary in same issue: 196(3): 601-601 in same issue.
**Highlighted in 2014 Genetics Spotlight booklet.
Remaining unanswered questions
Taken together, the data in this paper suggest that CaM Kinase II inhibits the regulated exocytosis DCVs and the subsequent release of neuropeptides from neuronal cell somas and dendrites. One major question that we were unable to definitively answer is Why do neurons inhibit the release of neuropeptides from cell somas and dendrites?
Fun factoids about this study
UNDER CONSTRUCTION
Related studies
UNDER CONSTRUCTION
