2005-2007: Discovery of the Missing Gq Effector Pathway
|
The Gq pathway is a critical part of the synaptic signaling network. It interacts closely with the other two G protein pathways in the network and is absolutely required for the growth and behavior of C. elegans (see Adventures>2000-2005B). Gq itself is highly conserved in all animals, having 82% identity between C. elegans and humans. In major Cell and Molecular Biology textbooks Gq is prominently featured in signal transduction chapters. In 2007, those textbooks informed us that Gq signals by activating Phospholipase C beta (PLC beta). PLC beta in turn converts the lipid PIP2 into the critical second messenger molecule Diacylglycerol (DAG).
Genetic studies in C. elegans are consistent with the above actions of the Gq pathway, but they also revealed that Gq signals through another unknown effector in neurons (see Adventures>1996-1999). We knew this because an animal with no Gq has a much stronger growth and locomotion phenotype than an animal with no PLC beta. |

|
The identity of the "missing" Gq effector pathway had been bugging my lab for years. We knew we could find it using the right forward genetic screen. In a forward genetic screen you randomly mutagenize animals, usually by exposing them to a chemical mutagen, and then screen their grandprogeny for mutations that disrupt the process you are studying (see Genetic Screens Explained). The mutations are then mapped to specific proteins. The beauty of forward genetic screens is that they are an unbiased way to let the organism tell you what genes/ proteins are important for the process you are studying.
The challenge we faced was to design a genetic screen that would reveal the missing Gq effector pathway. We finally settled on a strategy of screening for suppressors of a goa-1 (Go) null mutant. We and others had previously shown that GOA-1 (Go) negatively regulates the EGL-30 (Gq) pathway (see Adventures>1996-1999). In a goa-1 null mutant the excessive Gq pathway activity results in strongly hyperactive locomotion and slow growth. I reasoned that mutationally reducing the activity of one or more Gq effectors would restore normal levels of locomotion and improve the growth of the goa-1 null.
Meanwhile, Michael Ailion in Erik Jorgensen's lab had started a similar genetic screen. He screened for suppressors of an egl-30 gain-of-function mutant.
The challenge we faced was to design a genetic screen that would reveal the missing Gq effector pathway. We finally settled on a strategy of screening for suppressors of a goa-1 (Go) null mutant. We and others had previously shown that GOA-1 (Go) negatively regulates the EGL-30 (Gq) pathway (see Adventures>1996-1999). In a goa-1 null mutant the excessive Gq pathway activity results in strongly hyperactive locomotion and slow growth. I reasoned that mutationally reducing the activity of one or more Gq effectors would restore normal levels of locomotion and improve the growth of the goa-1 null.
Meanwhile, Michael Ailion in Erik Jorgensen's lab had started a similar genetic screen. He screened for suppressors of an egl-30 gain-of-function mutant.

|
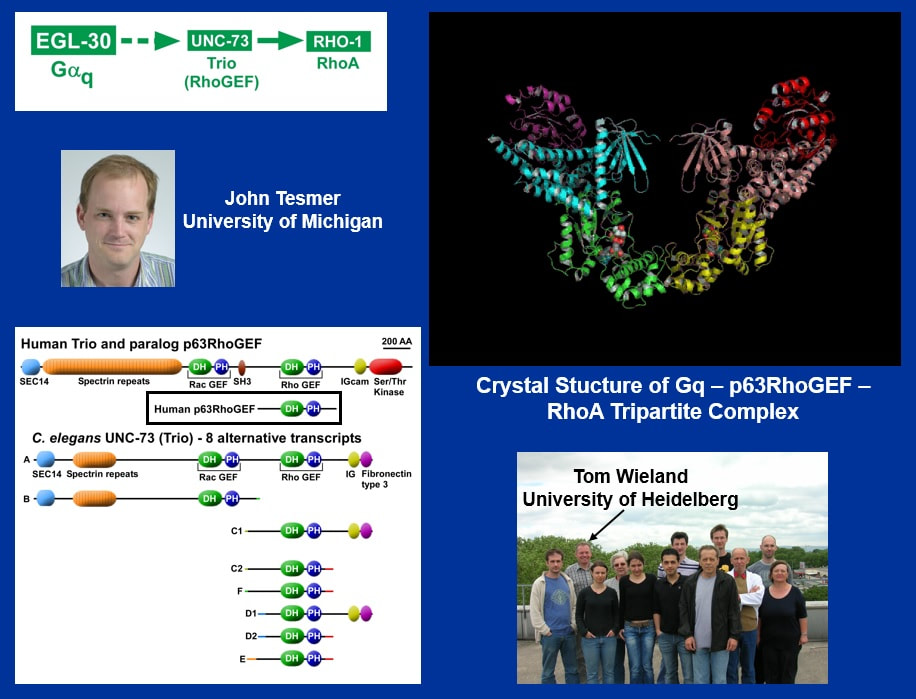
The Miller lab found 2 mutations, named ce362 and ce367, that mapped to the unc-73 gene. The Jorgensen lab also found 2 mutations that mapped to this gene. Pictures of one of the suppressor mutants are shown on the right. The unc-73 gene is large and complex with at least 8 alternative transcripts. The two major functional domains of the UNC-73 protein are the Rac GEF domain, which activates the Rac signal transduction protein and the Rho GEF domain, which activates the Rho signal transduction protein. |

|
The Rac GEF domain was known to be important for cell and neuronal axon migrations. The Rho GEF domain had not yet between assigned a specific function, but Rob Steven had recently isolated a mutation called ev802 that deleted the domain. All 4 of the combined Miller and Jorgensen lab mutations mapped to Rho GEF domain. Interestingly, C. elegans has the ability to make multiple short transcripts that lack the Rac GEF domain and only have the Rho GEF domain and some flanking sequences. Humans also have a gene called "p63 RhoGEF" that looks very similar to the UNC-73 Rho GEF protein. All of the Rho GEF mutations caused a slow/ sluggish locomotion rate.

|
Given UNC-73's prominent role in neuronal development, at least for the Rac GEF domain, we wanted to test if defects in neuronal development contributed to the sluggishness of unc-73 RhoGEF mutants. To do this, we expressed the unc-73e cDNA under control of an inducible heat shock promoter and induced its expression in adult unc-73(ce362) mutants, after neuronal development was complete. Under these conditions we were able to rescue the locomotion rate of unc-73(ce362) adults to greater than wild type levels, suggesting there were no significant developmental defects contributing to their sluggish locomotion. Animals can be sluggish due a muscle problem or a nervous system problem. To determine the extent to which neuronal defects contribute to the sluggishness of unc-73 mutants, we expressed the unc-73e cDNA pan-neuronally in unc-73(ce362) mutants. This transgene restored wild type locomotion to mutant. The above 2 experiments thus demonstrate that the sluggishness of UNC-73 RhoGEF domain mutants is caused by a defect in neuronal function and not neuronal development or muscle function. Our finding that unc-73 RhoGEF mutants suppress mutants with an overactive Gq pathway is consistent with UNC-73's RhoGEF domain acting as an effector in the Gq pathway, but we needed to further test this through double mutant analysis. If UNC-73's RhoGEF domain is the missing Gq effector, then a double mutant lacking both PLC beta and UNC-73 RhoGEF should have the appearance and phenotypes of a Gq null mutant. That is indeed what we found. Single mutants lacking only PLC beta or only UNC-73's RhoGEF domain were similarly sluggish, but not paralyzed. However, double mutants that were null for both proteins were almost completely paralyzed and had locomotion rates not significantly different from the egl-30 (Gq) null mutant. The doubles also resembled egl-30 (Gq) nulls in general appearance and growth characteristics. For example, due to lack of locomotion, newly hatched egl-30 nulls and unc-73 RhoGEF; PLC beta doubles ("double nulls") remained folded as they were in the egg for hours after hatching. egl-30 nulls and the double nulls also share a 100% early larval growth arrest phenotype, although the arrested larvae remain alive for a week or more after hatching. Using non-null unc-73 RhoGEF; PLC beta double mutants, we also showed that the synthetic interaction also extended to egg laying, with the double mutants laying their eggs at a decreased rate and becoming bloated with eggs comparable to strong reduction-of-function egl-30 mutants. We previously showed that the DAG analog phorbol myristate acetate, if mixed in the right doseage into the media of culture plates, could restore wild-type levels of locomotion to Gq null mutants within 2 hours (see Adventures>2000-2005B). If the double nulls are essentially the same as Gq nulls, then we should also be able to rescue their paralysis with this phorbol ester, and that is indeed the case. Within 2 hours of transferring the double null mutants to a plate with phorbol ester, their locomotion rate improved 360-fold, to a level greater than that of wild type animals on control plates. This result suggests that the double nulls are paralyzed for the same functional, non-developmental reason(s) as Gq nulls. In summary, the results so far are consistent with PLC beta and UNC-73's RhoGEF domain acting in separate pathways that together mediate the Gq signaling that drives the locomotion, growth, and egg laying of the animal. |





|
So, at this point we had taken the genetics about as far as we could go, but we were still left with an important question: Is Trio’s Rho-specific GEF domain a direct effector of Gq, or are there intermediate proteins in the pathway?
To address this, we needed a collaborator to do the biochemistry. As it turned out, the first person I contacted [click], John Tesmer, was working on a similar project in verterbrates. John is a structural biologist who studies interactions between G proteins and their effectors using X-ray crystallography.
John updated me on vertebrate biochemical studies that were pointing to the same conclusion as our C. elegans studies. In fact, John and his collaborator, Tom Wieland in Germany, were preparing a paper describing [click] the crystal structure of this complex of Gq bound to Rho through this Trio family protein called p63RhoGEF.
The problem with the vertebrate studies is that they lacked the genetics. They had no data on the physiological relevance of this new pathway in living animals, so they were very excited to hear about our C. elegans studies.
In fact, they were so excited that they collaborated with us and showed us that the biochemistry of the C. elegans proteins is the same as the biochemistry of the vertebrate proteins, and that our C. elegans mutations, when reproduced on p63RhoGEF, strongly impair the ability of Gq to activate RhoA.
To address this, we needed a collaborator to do the biochemistry. As it turned out, the first person I contacted [click], John Tesmer, was working on a similar project in verterbrates. John is a structural biologist who studies interactions between G proteins and their effectors using X-ray crystallography.
John updated me on vertebrate biochemical studies that were pointing to the same conclusion as our C. elegans studies. In fact, John and his collaborator, Tom Wieland in Germany, were preparing a paper describing [click] the crystal structure of this complex of Gq bound to Rho through this Trio family protein called p63RhoGEF.
The problem with the vertebrate studies is that they lacked the genetics. They had no data on the physiological relevance of this new pathway in living animals, so they were very excited to hear about our C. elegans studies.
In fact, they were so excited that they collaborated with us and showed us that the biochemistry of the C. elegans proteins is the same as the biochemistry of the vertebrate proteins, and that our C. elegans mutations, when reproduced on p63RhoGEF, strongly impair the ability of Gq to activate RhoA.
|
So, complementary approaches ranging from the genetics of living animals down to the structure of molecular complexes had converged on an important conclusion: Trio’s Rho-specific GEF domain is the missing Gq effector.
|

|
Williams SL, Lutz S, Charlie NK, Vettel C, Ailion M, Coco C, Tesmer JJ, Jorgensen EM, Wieland T, and Miller KG (2007). Trio’s Rho – Specific GEF domain is the missing Gq Effector in C. elegans. Genes Dev. 21:2731-2746. PMCID: PMC2045128. *
*Featured article. See journal commissioned Perspective.
Cited by 50 papers as of April, 2018.
*Featured article. See journal commissioned Perspective.
Cited by 50 papers as of April, 2018.
Remaining Important Questions
We are intrigued by our finding that phorbol esters acutely rescue the locomotion of both the egl-30 (Gq) null and the unc-73 RhoGEF; egl-8 PLC beta double null mutants to similar degrees. Since phorbol ester is a DAG analog this could simply indicate that overactivation of the PLC-beta (DAG producing) branch can compensate for lack of a Trio RhoGEF effector branch. Alternatively, this result could indicate that the Trio RhoGEF branch ultimately regulates DAG levels as well. In support of this second possibility, mammalian RhoA has been shown to bind and inhibit DAG Kinase (known as DGK-1 in C. elegans). The C. elegans DAG Kinase ortholog DGK-1 has also been shown to bind to the C. elegans RhoA ortholog (McMullan et al., 2006) and is a component of the Gq signaling pathway in neurons (see Adventures>1996-1999).
DAG Kinase normally breaks down DAG. Thus, knocking out the RhoA pathway should cause an overactive DAG Kinase and a reduction in DAG levels. The synthetic effect of Trio RhoGEF/ PLC-beta double mutants may thus be due to an inability to synthesize DAG from the Gq-PLC beta pathway coupled with an inability to prevent the breakdown of DAG as mediated by the Gq-Trio RhoGEF pathway.
A fourth G-protein signaling pathway, G12, has also been shown to activate RhoA in mammals and C. elegans. However, knocking out this pathway has no effect on locomotion rate or aldicarb sensitivity in C. elegans (Hiley et al., 2006). This could indicate that upregulation of either or both branches of the Gq pathway can compensate for lack of the G12 pathway; however, the reverse is not true since Gq null mutants or Trio-PLC beta double nulls are completely paralyzed and larval arrested.
Finally, in C. elegans, The Gq-Trio-RhoA pathway is also known to exert its effects through the generation of the small signaling molecule sphingosine-1-phosphate by sphingosine kinase (Chan et al., 2006). This is a DAG-independent mechanism (Chan et al., 2006). Although this pathway has been shown to have effects on the appearance of SV clusters and on the localization of the synaptic protein UNC-13, it has very little effect on the behavior or growth of animals. Future studies need to test the in vivo relevance of this pathway, and its relative contributions to Gq-Trio-RhoA signaling, by making sphingosine kinase/ PLC-beta double mutants. If it is a major route for Gq-Trio-RhoA signaling, then doubles with PLC-beta should look similar Trio RhoGEF/ PLC-beta double nulls (i.e. larval arrested and paralyzed).
DAG Kinase normally breaks down DAG. Thus, knocking out the RhoA pathway should cause an overactive DAG Kinase and a reduction in DAG levels. The synthetic effect of Trio RhoGEF/ PLC-beta double mutants may thus be due to an inability to synthesize DAG from the Gq-PLC beta pathway coupled with an inability to prevent the breakdown of DAG as mediated by the Gq-Trio RhoGEF pathway.
A fourth G-protein signaling pathway, G12, has also been shown to activate RhoA in mammals and C. elegans. However, knocking out this pathway has no effect on locomotion rate or aldicarb sensitivity in C. elegans (Hiley et al., 2006). This could indicate that upregulation of either or both branches of the Gq pathway can compensate for lack of the G12 pathway; however, the reverse is not true since Gq null mutants or Trio-PLC beta double nulls are completely paralyzed and larval arrested.
Finally, in C. elegans, The Gq-Trio-RhoA pathway is also known to exert its effects through the generation of the small signaling molecule sphingosine-1-phosphate by sphingosine kinase (Chan et al., 2006). This is a DAG-independent mechanism (Chan et al., 2006). Although this pathway has been shown to have effects on the appearance of SV clusters and on the localization of the synaptic protein UNC-13, it has very little effect on the behavior or growth of animals. Future studies need to test the in vivo relevance of this pathway, and its relative contributions to Gq-Trio-RhoA signaling, by making sphingosine kinase/ PLC-beta double mutants. If it is a major route for Gq-Trio-RhoA signaling, then doubles with PLC-beta should look similar Trio RhoGEF/ PLC-beta double nulls (i.e. larval arrested and paralyzed).
Fun Factoids about this Study
-Our study showed that the Trio RhoGEF pathway is the dominant Gq effector branch for driving growth and egg laying, whereas the two pathways make about equal contributions to the locomotion behavior.
-We tried to find out which pathway came first in evolution. However, we found orthologs of all Gq, PLC-beta, and Trio RhoGEF in the sea anemone Nematostella, indicating that Gq may already have been signaling through both PLC-beta and Trio RhoGEF in the last common cnidarian-bilaterian animal ancestor, which lived an estimated 670-820 million years ago.
-I gave a talk on this work as an invited speaker at the 2007 G protein Signaling Gordon Conference in Biddeford, Maine.
-We tried to find out which pathway came first in evolution. However, we found orthologs of all Gq, PLC-beta, and Trio RhoGEF in the sea anemone Nematostella, indicating that Gq may already have been signaling through both PLC-beta and Trio RhoGEF in the last common cnidarian-bilaterian animal ancestor, which lived an estimated 670-820 million years ago.
-I gave a talk on this work as an invited speaker at the 2007 G protein Signaling Gordon Conference in Biddeford, Maine.
Related Studies
Concomitant with the discovery of Trio RhoGEF as a Gq effector in C. elegans, biochemical and structural studies demonstrated that mammalian Gq binds to and activates the orthologous mammalian Rho GEF proteins. Thus, this turned out to be a timely study, represented by 3 papers in 2007. The unique aspect of our paper was that it provided genetic data on the biological significance of this new pathway in a living animal.
Lutz, S., Shankaranarayanan, A., Coco, C., Ridilla, M., Nance, M.R., Vettel, C., Baltus, D., Evelyn, C.R., Neubig, R.R., Wieland, T., et al. (2007). Structure of Gq-p63RhoGEF-RhoA complex reveals a pathway for the activation of RhoA by GPCRs. Science 318, 1923-1927.
Rojas, R.J., Yohe, M.E., Gershburg, S., Kawano, T., Kozasa, T., and Sondek, J. (2007). Gq directly activates p63RhoGEF and Trio via a conserved extension of the Dbl homology-associated pleckstrin homology domain. J Biol Chem 282, 29201-29210.
Williams, S.L., Lutz, S., Charlie, N.K., Vettel, C., Ailion, M., Coco, C., Tesmer, J.J., Jorgensen, E.M., Wieland, T., and Miller, K.G. (2007). Trio's Rho-specific GEF domain is the missing Gq effector in C. elegans. Genes Dev 21, 2731-2746.
As a postdoc in Tony Pawson's lab, Rob Steven produced a mutation that was crucial to our study: the ev802 mutation that is an in-frame deletion of RhoGEF domain of Trio and a null mutant for Trio's RhoGEF domain.
Steven, R., Zhang, L., Culotti, J., and Pawson, T. 2005. The UNC-73/Trio RhoGEF-2 domain is required in separate isoforms for the regulation of pharynx pumping and normal neurotransmission in C. elegans. Genes Dev 19(17): 2016-2029.
Stephen Nurrish's lab at University College in London published important findings about RHO-1 binding to DGK-1 and provided genetic results consistent with RHO-1 inhibiting DGK-1. He also provided the first genetic studies of the G12-p115RhoGEF-RHO-1 pathway in C. elegans.
McMullan, R., Hiley, E., Morrison, P., and Nurrish, S.J. 2006. Rho is a presynaptic activator of neurotransmitter release at pre-existing synapses in C. elegans. Genes Dev 20(1): 65-76.
Hiley, E., McMullan, R., and Nurrish, S.J. 2006. The Ga12-RGS RhoGEF-RhoA signalling pathway regulates neurotransmitter release in C. elegans. Embo J 25(24): 5884-5895.
Derek Sieburth's lab provided the first characterization of the sphingosine kinase that appears to function downstream of RHO-1 in C. elegans.
Chan, J.P., Hu, Z., and Sieburth, D. (2012). Recruitment of sphingosine kinase to presynaptic terminals by a conserved muscarinic signaling pathway promotes neurotransmitter release. Genes Dev. 26, 1070-1085.
Lutz, S., Shankaranarayanan, A., Coco, C., Ridilla, M., Nance, M.R., Vettel, C., Baltus, D., Evelyn, C.R., Neubig, R.R., Wieland, T., et al. (2007). Structure of Gq-p63RhoGEF-RhoA complex reveals a pathway for the activation of RhoA by GPCRs. Science 318, 1923-1927.
Rojas, R.J., Yohe, M.E., Gershburg, S., Kawano, T., Kozasa, T., and Sondek, J. (2007). Gq directly activates p63RhoGEF and Trio via a conserved extension of the Dbl homology-associated pleckstrin homology domain. J Biol Chem 282, 29201-29210.
Williams, S.L., Lutz, S., Charlie, N.K., Vettel, C., Ailion, M., Coco, C., Tesmer, J.J., Jorgensen, E.M., Wieland, T., and Miller, K.G. (2007). Trio's Rho-specific GEF domain is the missing Gq effector in C. elegans. Genes Dev 21, 2731-2746.
As a postdoc in Tony Pawson's lab, Rob Steven produced a mutation that was crucial to our study: the ev802 mutation that is an in-frame deletion of RhoGEF domain of Trio and a null mutant for Trio's RhoGEF domain.
Steven, R., Zhang, L., Culotti, J., and Pawson, T. 2005. The UNC-73/Trio RhoGEF-2 domain is required in separate isoforms for the regulation of pharynx pumping and normal neurotransmission in C. elegans. Genes Dev 19(17): 2016-2029.
Stephen Nurrish's lab at University College in London published important findings about RHO-1 binding to DGK-1 and provided genetic results consistent with RHO-1 inhibiting DGK-1. He also provided the first genetic studies of the G12-p115RhoGEF-RHO-1 pathway in C. elegans.
McMullan, R., Hiley, E., Morrison, P., and Nurrish, S.J. 2006. Rho is a presynaptic activator of neurotransmitter release at pre-existing synapses in C. elegans. Genes Dev 20(1): 65-76.
Hiley, E., McMullan, R., and Nurrish, S.J. 2006. The Ga12-RGS RhoGEF-RhoA signalling pathway regulates neurotransmitter release in C. elegans. Embo J 25(24): 5884-5895.
Derek Sieburth's lab provided the first characterization of the sphingosine kinase that appears to function downstream of RHO-1 in C. elegans.
Chan, J.P., Hu, Z., and Sieburth, D. (2012). Recruitment of sphingosine kinase to presynaptic terminals by a conserved muscarinic signaling pathway promotes neurotransmitter release. Genes Dev. 26, 1070-1085.
